Summary
Background
The 2013–16 Ebola virus disease epidemic in west Africa caused international alarm due to its rapid and extensive spread resulting in a significant death toll and social unrest within the affected region. The large number of cases provided an opportunity to study the long-term kinetics of Zaire ebolavirus-specific immune response of survivors in addition to known contacts of those infected with the virus.
Methods
In this observational cohort study, we worked with leaders of Ebola virus disease survivor associations in two regions of Guinea, Guéckédou and Coyah, to recruit survivors of Ebola virus disease, contacts from households of individuals known to have had Ebola virus disease, and individuals who were not knowingly associated with infected individuals or had not had Ebola virus disease symptoms to serve as negative controls. We did Zaire ebolavirus glycoprotein-specific T cell analysis on peripheral blood mononuclear cells (PBMCs) on location in Guinea and transported plasma and PBMCs back to Europe for antibody quantification by ELISA, functional neutralising antibody analysis using live Zaire ebolavirus, and T cell phenotype studies. We report on the longitudinal cellular and humoral response among Ebola virus disease survivors and highlight potentially paucisymptomatic infection.
Findings
We recruited 117 survivors of Ebola virus disease, 66 contacts, and 23 negative controls. The mean neutralising antibody titre among the Ebola virus disease survivors 3–14 months after infection was 1/174 (95% CI 1/136—1/223). Individual results varied greatly from 1/10 to more than 1/1000 but were on average ten times greater than that induced after 1 month by single dose Ebola virus vaccines. Following reactivation with glycoprotein peptide, the mean T cell responses among 116 Ebola virus disease survivors as measured by ELISpot was 305 spot-forming units (95% CI 257–353). The dominant CD8+ polyfunctional T cell phenotype, as measured among 53 Ebola virus disease survivors, was interferon γ+, tumour necrosis factor+, interleukin-2–, and the mean response was 0·046% of total CD8+ T cells (95% CI 0·021–0·071). Additionally, both neutralising antibody and T cell responses were detected in six (9%) of 66 Ebola virus disease contacts. We also noted that four (3%) of 117 individuals with Ebola virus disease infections did not have circulating Ebola virus-specific antibodies 3 months after infection.
Interpretation
The continuous high titre of neutralising antibodies and increased T cell response might support the concept of long-term protective immunity in survivors. The existence of antibody and T cell responses in contacts of individuals with Ebola virus disease adds further evidence to the existence of sub-clinical Ebola virus infection.
Funding
US Food & Drug Administration, Horizon 2020 EU EVIDENT, Wellcome, UK Department for International Development.
Introduction
,
,
,
Vaccine studies in non-human primates also suggest that CD8+, interferon (IFN)+, tumour necrosis factor+, interleukin 2 (IL-2)–/+ T cells are a potential correlate of protection.
,
The Ebola virus vaccine, Ervebo, which is based on the viral glycoprotein, was evaluated during the 2013–16 epidemic. Ring vaccination trials estimated vaccine efficacy to be 100%.
Previous studies on survivor Ebola virus disease immunology have been done but only antibody levels were monitored. During historical Ebola virus outbreaks, plasma from survivors had been used to treat individuals suffering from acute Ebola virus disease; however, a convalescent plasma therapy trial in west Africa did not show significant clinical benefit or a correlation between neutralisation and efficacy.
These results suggest that both antibody and T cell responses might have a role in protection in humans.
Evidence before this study
We searched the PubMed database with MESH terms “Ebola”, “immunity”, “sero-prevalence”, “paucisymptomatic”, and for articles published between database inception and April 15, 2020, in any language. There was one report on long-term antibody and T cell responses in 11 survivors split across two small Sudan ebolavirus outbreaks. Neutralisation assays revealed titres of 0–1/80, and T cell proliferation and kinetics analyses clearly showed CD8+ T cell activation, but quantification by ELISpot was not done. A comprehensive analysis of ELISA responses in survivors from the 1995 Kikwit outbreak showed antibody titres plateaued at 3 weeks to 1 year after onset of symptoms. A report by Leroy and colleagues revealed the existence of paucisymptomatic individuals with Ebola virus infections. Additionally, work by Glynn and colleagues further demonstrates the presence of asymptomatic Ebola virus infection. Several reports showed persistence of Ebola virus in immune privileged sites, and an in-depth longitudinal analysis of the B cell response among four Ebola virus disease survivors has also been done. We found no other studies reporting the longitudinal analysis of cellular and humoral immunity among survivors over 3 consecutive years.
Added value of this study
This unique study reports an in-depth analysis of naturally acquired immunity to Ebola virus and enables a comprehensive comparison between naturally acquired and vaccine-induced immunity to Ebola virus both at the antibody and T cell level. The study also provides supporting evidence for the existence of paucisymptomatic Ebola virus disease and suggests that true incidence of Ebola virus infection in the west African outbreak was greater than recorded. Additionally, T cell phenotyping results support the preclinical findings of a potential correlate of protection to Ebola virus.
Implications of all the available evidence
We suggest that up to 9% of individuals with Ebola virus diseaese will present with mild symptoms, which will have implications with regard to monitoring and responding to future outbreaks. Additionally, the work here shows that T cell responses are relevant and long lasting among survivors of Ebola virus disease; therefore, future vaccine developers might wish to consider the T-cell response in more depth. The evaluation of convalescent plasma showed varying titres of anti-Ebola virus IgG, so pre-screening of IgG and neutralising titres before administration of convalescent plasma should be considered in future outbreaks.
Similar observations were also reported in the 2013–16 west African epidemic.
Such observations are rarely reported in detail and do not include in-depth immunology or molecular diagnostic data. However, a case report
with associated sequence data suggests that a paucisymptomatic mother was able to transmit Ebola virus to her baby. A better understanding of the incidence of paucisymptomatic Ebola virus disease is needed and might have implications in assessing future transmission risks. Additionally, systematic reviews
,
of sero-epidemiology studies of populations in Africa reported that about 8% of individuals had antibodies specific to Ebola virus and other related members of the filovirus family. The absence of reported Ebola virus disease outbreaks in such populations is puzzling but could be explained by antibody cross reactivity, sub-clinical Ebola virus disease or increased rates of infectious diseases in general.
A complete understanding of naturally acquired immunity to Ebola virus will help improve the design and evaluation of experimental vaccines, convalescent plasma, and therapeutic antibodies. Assessment of the memory cell phenotype in survivors of Ebola virus disease might also provide insight into chronic viral shedding from immune privileged sites. Furthermore, assessment of the immune response of individuals who are not known to be Ebola virus-positive but exposed to Ebola virus disease might reveal the extent of paucisymptomatic infections and help in the assessment of the incidence of Ebola virus infection and the associated case fatality rate.
Methods
Study designs and participants
Procedures
After 18–20 h incubation at 37°C, IFNγ release was determined by standard ELISpot protocol (Mabtech; Nacka Strand, Sweden) and spot forming cells enumerated using an S6 core analyser (Cellular Technology; Shaker Heights, OH). IFNγ release was calculated by subtracting the background from each well and taking the mean of three triplicate wells. The results were determined as spot forming units per one million cells and IFNγ response to the Ebola virus glycoprotein peptide were summed to determine the overall T-cell response.
Briefly, high binding microtitre plates were coated with whole Ebola virus inactivated virions and incubated for 16–20 h. After washing in PBS and 0·1% Tween20 (PBST) and blocking (PBS and 5% milk powder), 1/200 dilutions of plasma sample were added to the plates and incubated for 1 h. Horseradish peroxidase-conjugated polyclonal antibody (P0214, Dako; Santa Clara, CA; dilution 1/1000) in conjunction with TMB substrate was used to develop the reaction. Optical density was determined at 450 nm minus 630 nm (reference wavelength). Each sample was analysed in duplicate on mock and viral antigen. The mean optical density of each sample on the mock antigen was subtracted from the mean optical density of the respective sample on the Ebola virus antigen. Arbitrary ELISA units were extrapolated by linear regression analysis using standard curves generated from patient antiserum. Further specificity was assessed using a Zaire, Makona Ebola virus glycoprotein (sourced from Nuffield Department of Medicine, Oxford University, Oxford, UK) specific ELISA, for which Nunc Maxisorb 96-well plates (Merck; Darmstadt, Germany) were coated overnight (16–18 h) with purified Ebola virus glycoprotein antigen (0·5 μg/mL). Plasma was serially diluted, starting at 1/200 and the bound IgG was detected using goat anti-human IgG Fcγ specific antibody conjugated to alkaline phosphatase (1/15000). Alkaline phosphatase-yellow substrate (Sigma) was added and the optical density measured at 405 nm using a VERSAmax plate reader (Molecular Devices; Wokingham, UK) controlled by SoftMax Pro Enterprise software (version 4.7.1). The plates were read using a predefined Softmax template, which fits a four-parameter logistic curve to the dose response data. The cutoff was defined as the mean negative value plus five SDs.
Briefly, after heat treatment for complement inactivation, plasma was serially diluted in supplemented Dulbecco’s modified Eagle’s medium (DMEM) in 96-well culture plates, 100 median tissue culture infectious dose (TCID50) units of Ebola virus variant Mayinga were added to the plasma dilutions. Following incubation at 37°C for 1 h, Vero cell suspension in supplemented DMEM was added. Plates were then incubated at 37°C with 5% CO2 and cytopathic effects were evaluated at 7 days after infection. Neutralisation titres were calculated as geometric mean titre of four replicates. A titre of 1/8 or above is classified as positive.
Recombinant glycoprotein, nucleoprotein, and viral protein 35 were generated based on the Ebola virus strain Makona in HEK293T cells and whole cell lysates were used. Viral protein 40 was based on Ebola virus strain Kikwit and was obtained from Stratech Scientific (Ely, UK). Proteins were heat denatured and loaded onto 4–12% BisTris gels and separated by size by SDS-PAGE. The proteins were then transferred to polyvinylidene difluoride membrane and blocked overnight in block buffer (PBST buffer with 5% milk). Plasma was diluted 1/1000 in block buffer and incubated with the Ebola virus-protein containing blots for 4 h at room temperature. The blots were washed for 5 min in PBST. Secondary antibody (goat anti-human IgG [γ-chain specific] peroxidase conjugate) F(ab’)2 fragments (Sigma; A2290), were diluted 1/1000 in block buffer. The blots were incubated with secondary antibody for 1 h at room temperature. Membranes were washed and blots developed with ECL prime, incubating for 5 min. Images were captured at 5 min and 10 min exposure and presence of immunoreactivity determined against a molecular marker standard.
Briefly, PBMCs were resuspended in warmed complete media (Roswell Park Memorial Institute medium supplemented with penicillin–streptomycin, fetal calf serum, L-glutamine, HEPES, and 2-mercaptoethanol) and rested overnight at 37°C. The following day cells were adjusted to 1 × 106 cells/mL in media containing anti-CD28 BUV737, CD49d, and CD107a-PerCP cy5.5 (1 μg/mL). Samples were then left either untreated or were stimulated with an Ebola virus glycoprotein peptide pool, containing 187 15mer overlapping peptides at 2·5 μg/peptide or 1 μg/mL staphylococcal enterotoxin B for 16–18 h, as previously described.
,
2 h into the incubation, brefeldin A and monensin (1 μg/mL) were added to block cytokine secretion from the cell. The following day samples were washed in cold FACS wash and LIVE/DEAD fixable aqua dye (Life Technologies; Carlsbad, CA) was added. Samples were washed, then incubated with a cell surface cocktail of antibodies including CD3-APC 750, CD4-BV786, CD8-AF700, CD19-BV510, CD14-BV510, CCR7-APC, CD95-BUV395, and CD45RO-BV605. Cells were then washed, fixed and permeabilised using Becton Dickinson (London, UK) Cytofix/Cytoperm, and stained for intracellular cytokines using IFNγ-AF488, TNF-BV421, and IL-2-PE. Samples were then washed, resuspended, and acquired using a Becton Dickinson Fortessa machine and FACS Diva software. Sample analysis used FlowJo, Pestle, and SPICE software as described previously.
All antibodies were obtained from Biolegend (London, UK), with the exception of CD95-BUV395 and CD28 BUV737, which were obtained from Becton Dickinson.
Statistical analysis
The data collected from all the volunteers were categorised into three groups, survivor, contact, and negative, which were sub-divided by region and sex. Measurements for ELISpot, ELISA and neutralisation were tested independently. Statistical analysis on the fixed effect coefficient for year was done using R version 4.0.1 with the lme4 package version 1.1–23 and fitted models were assessed for violation of assumptions. Cytokine responses were determined by subtracting the untreated response from that of the stimulation; negative values were set to 0·001 and statistical differences were determined using Mann-Whitney test. Correlations were determined using Spearman correlation analysis. Statistical tests were done using GraphPad Prism version 8.
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
TableNumbers of participants from Coyah and Guéckédou sample sites

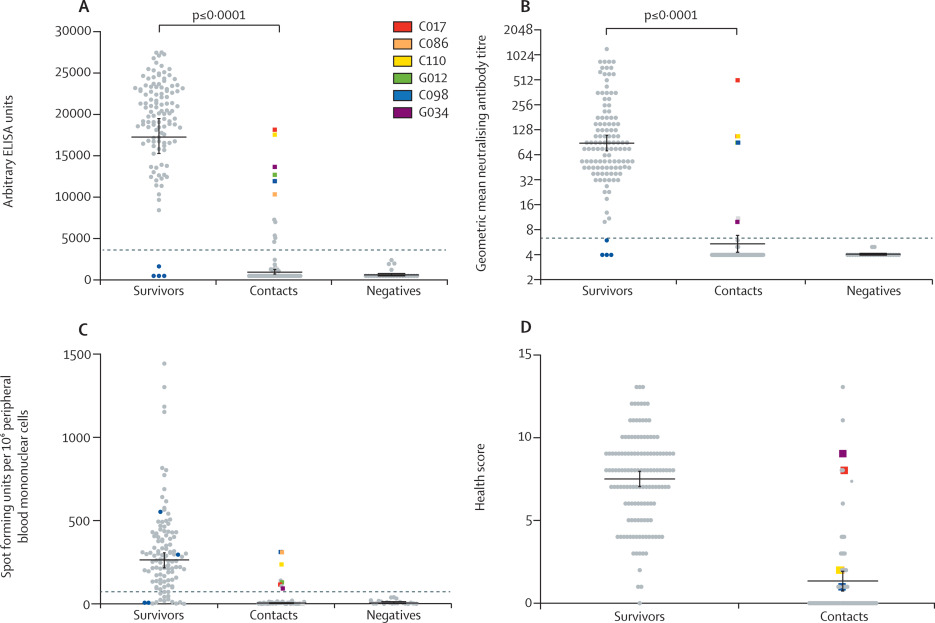
Figure 1Characterisation of immune responses of Ebola virus disease survivors and close contacts
(A) Ebola virus-specific antibody titre against whole virus preparation as measured by ELISA in plasma samples collected from survivors (n=117), contacts (n=66), and negative controls (n=23). (B) Geometric mean neutralising antibody titre against live Ebola virus (strain Mayinga) using plasma samples collected from survivors (n=117), contacts (n=66), and negative controls (n=23). Results displayed on a log2 scale. (C) ELISpot to detect IFNγ secretion using an Ebola virus glycoprotein peptide library and peripheral blood mononuclear cell samples from survivors (n=116), contacts (n=42), and negative controls (n=21). (D) Quantified results from health questionnaire given to all survivors and contacts. A score of 13 represents severe symptoms and 0 represents no symptoms during the 2013–16 outbreak. Bars represent the geometric mean plus or minus the standard error of the mean. Dashed lines represent the mean plus five SDs. Mann-Whitney tests were done for all statistical analyses. Blue dots represent seronegative survivors. Coloured squares represent six (9%) of the 66 contacts who had Ebola virus-specific neutralising antibodies. IFNγ=interferon γ.

Figure 2Longitudinal response following Ebola virus infection
(A) Ebola virus-specific antibody titre against whole virus preparation as measured by ELISA in plasma samples collected from matched survivors (n=96) in 2015–17. (B) Neutralising antibody titre against live virus preparation (Mayinga) using plasma samples collected from matched survivors (n=96) in 2015–17. (C) ELISpot to detect IFNγ secretion using an Ebola virus glycoprotein peptide library and peripheral blood mononuclear cell samples from matched survivors (n=63) in 2015–17. (D) Time in months from when each volunteer was confirmed Ebola virus-negative following second negative PCR and so were discharged from the Ebola treatment centre. Data include antibody titre to whole Ebola virus as determined by ELISA, antibody activity as determined by neutralisation assay, and T-cell responses as determined by ELISpot. Parts A–C show the geometric mean with 95% CI. IFNγ=interferon γ.

Figure 3T cell activation in response to Ebola virus glycoprotein peptide
Samples collected from 2016 were used to analyse the T cell response to Ebola virus disease. (A) The sum of CD8+ T cells producing IFNγ in response to overnight stimulation with glycoprotein peptides (2·5 μg per peptide per mL). Bars represent the geometric mean plus or minus the standard error of the mean. (B) Representative dot plot of CD8+ T cell phenotype. IFNγ producing cells are highlighted in red. (C) The sum of CD4+ T cells producing IFNγ in response to overnight stimulation with glycoprotein peptides (2·5 μg per peptide per mL). Bars represent the geometric mean ± the standard error of the mean. (D) Representative dot plot of the CD4+ T cell phenotype. IFNγ producing cells are highlighted in red. IFNγ=interferon γ.
suggested a role for so called polyfunctional CD8+ T cells, which produce multiple cytokines, in the control of Ebola virus disease; we therefore investigated the incidence of IFNγ+, TNF+ or IFNγ+, TNF+, IL-2+ CD8+ T cells among 53 survivor PBMC samples collected in 2016 (15–28 months after infection). When these PBMCs were stimulated overnight with Ebola virus glycoprotein peptide pools, a significant proportion of T cells were IFNγ+, TNF+, and CD8+ compared with 26 contacts who were seronegative by ELISA and neutralisation (p=0·0004; figure 4). Significant populations of IFNγ+ (p=0·018) only and IFNγ+, TNF+ (p=0·0075) CD8+ T cells were identified among the survivors’ PBMCs compared with ELISpot negative controls. Survivor samples had a significantly larger proportion of CD107a expressing cells than their comparative contacts (p=0·032), and when backgated, the majority of these CD107a+ cells were IFNγ+, TNF+, CD8+ T cells (figure 4B). CD8+ T cells originating from survivors of Ebola virus disease showed significantly greater amounts of CD107a than negative controls (p=0·032). There was a strong correlation between total Ebola virus ELISA and neutralisation assay results (appendix 2, p 8). We also observed a significant correlation between ELISpot and ELISA as well as ELISpot and neutralisation results (appendix 2, p 8). We did not find a significant correlation or difference between either age, sex, or virus load at time of diagnosis (cycle threshold values) and any of the immunological parameters measured (appendix 2, pp 8–9).

Figure 4Polyfunctional CD8+ T cell analysis
(A) Cytokines secreted by CD8+ T cells. Survivors’ PBMCs are represented by red dots and ELISpot negative controls by grey dots. Samples that returned a negative result during analysis were assigned a value of 0·001. (B) CD8+ T cell activation as measured by intracellular staining for the degranulation marker CD107a. (C) Representative dot plot of survivor PBMCs stimulated with Ebola virus glycoprotein peptide. CD107a+ events are highlighted in red. PBMC=peripheral blood mononuclear cell. IL-2=interleukin 2. IFNγ=interferon γ. TNF=tumour necrosis factor.
Discussion
,
. This finding is compelling evidence for long-term protection against reinfection with Ebola virus and bodes well for survivors if the disease returns to west Africa, suggesting that they could continue to have a role in front-line activities to control future outbreaks. However, the absence of humoral immunity in a small percentage of survivors might explain observations that a minority of Ebola virus disease survivors can be reinfected, suggesting it would be essential to assess their immunity before the risk of potential re-exposure. Of the four survivors who displayed no antibody response, two showed a detectable ELISpot response; however, this response was not present in subsequent samples and misuse of survivor certificates cannot be ruled out in this instance. Absence of antibody responses has been documented for various emerging diseases, including severe acute respiratory syndrome coronavirus 2 antibody negative samples from COVID-19 convalescent individuals.
From these data we would suggest that sero-epidemiology studies for Ebola virus might underestimate the prevalence of the disease. The disparity between vaccinees and survivors also highlights the need to periodically boost vaccine immunity or use alternative heterologous vaccine vector approaches that might induce a more durable response. However, it is yet to be determined what constitutes a protective antibody titre. The continuously high titres of neutralising antibody and T cell responses are perhaps unsurprising, because Ebola virus has been reported to persist for more than 1 year in immune privileged sites.
,
,
Furthermore, where we have found responses to be stable up to 3 years after infection, studies of survivors from Ebola virus disease outbreaks suggest that ELISA antibody responses continue to be relatively stable for more than 1 year and that low titres of neutralising antibodies have been found in survivors 40 years after infection.
,
However, further work is needed to show antigen persistence in these survivor samples.
Obvious study differences, including an absence of longitudinal sampling and of experimental treatment, might explain the differing results. Of note, the longitudinal analysis in our study uses only data from participants who provided a full 3 years’ worth of data; therefore, data from participants with missing or incomplete data was discarded, which is a limitation of this study with regard to the stability over time. Long-term, potent Ebola virus responses could be a result of continuous exposure to viral antigen as it seeps back into the systemic environment from immune privileged locations. There is evidence that relapse and transmission can occur long after the primary infection has been resolved,
and it would be of great interest to sample such immune privileged sites from our cohorts to see if there is live virus present. However, such an approach was not possible owing to biosafety constraints and the limitations of our ethical approval. We suggest that our studies do not support the presence of recirculating antigen 15 months or more after recovery, because we believe that CD8+ T cell phenotyping revealed a possible subset of T memory stem cells. T memory stem cells, are a self-renewing population of lymphocytes that are CD45RO–, CCR7+, CD27+, and CD95+.
In support of this suggestion, the phenotype of IFNγ-producing CD4+ T cells was CD45RO+ (figure 3D), suggesting that these cells are memory T cells and not terminally differentiated effectors that have recently been exposed to antigen. LaVergne et al
found that post Ebola syndrome was associated with greater activation of CD4+ and CD8+ T cells, so it would be of interest to correlate the magnitude of our T cell responses with post Ebola syndrome in any follow up studies.
However, the 2013–16 outbreak enabled a more comprehensive study to be done, which reported an absence of statistically significant improvement in survival associated with plasma treatment.
In the same trial, neutralising antibody titres of plasma harvested from Ebola virus disease survivors were on average ten times smaller than seen in our study. However, differences between assay conditions could account for this outcome, which might have affected the subsequent analysis of functional antibody activity and clinical outcome.
Furthermore, Sahr et al
showed a positive effect with plasma therapy in a small clinical trial in Sierra Leone.
However, our seropositive contacts showed both neutralising antibody and T cell responses, and we were able to show that five individuals also had antibodies specific to Ebola virus nucleoprotein, viral protein 35, and viral protein 40, thus confirming that they must have come into contact with the virus. The extent to which these contacts were symptomatic remains unknown and is difficult to address after the event and largely relies on anecdotal evidence. Matters are further complicated owing to the stigma surrounding Ebola virus disease in west Africa, meaning that individuals who were infected at the time were likely to have downplayed their symptoms or not sought treatment. However, owing to the health questioning and interviews that took place we are confident that the paucisymptomatic contacts within our cohort did have mild disease that did not prevent them from conducting their normal duties. Therefore, these observations strongly suggest that four potential outcomes from Ebola virus exposure exist: symptomatic infection resulting in death, symptomatic infection with survival and immunity, paucisymptomatic infection with survival and immunity, and no infection and no immunity. Additional support for the existence and potential transmission from paucisymptomatic people comes from a report whereby a mother was able to transmit Ebola virus to her child through breastmilk. Both parents reported no symptoms of Ebola virus disease and blood samples were PCR negative; however, semen and breast milk samples were shown to be PCR positive, and breastfeeding was concluded to be the most likely route of transmission.
Furthermore, a number of serological studies have suggested the existence of paucisymptomatic infection in west Africa.
,
It might not be possible to categorically show that these positive contacts were paucisymptomatic or showed mild disease; however, this study and others
,
suggest that this aspect of Ebola virus transmission and the role it might have in spreading Ebola virus disease needs to be considered during any future outbreaks. Additionally, the number of individuals infected with Ebola virus could be significantly larger than the officially reported numbers, which will also affect the extent of the reported mortality rate.
The importance of this T cell activity to Ebola virus vaccine design has been the subject of debate. The ChAd3, MVA boost regime has been shown to induce a strong T cell response and primate studies have revealed that CD8+ polyfunctional T cells might have a key role in protection from Ebola virus challenge, following vaccination with an adeno vaccine vector.
Our results show that the IFNγ+ and TNF+ double-positive CD8+ T cell population, which is known to be involved in long-term protection in primates, is also the dominant CD8+ T cell population in responses to restimulation with glycoprotein peptide in Ebola virus disease survivors. Therefore, further work should be done to determine the activity and proportion of CD8+ polyfunctional T cells in response to Ebola virus vaccination.
Contributors
PM, M3C, SG, and MKK conceived the study and experimental design with input from GB, MM, LL, MG, DP, OS, LK, EN, SB, HR, JH, AMH-R, and NM. Sample collection within Guinea was led by JAB and MKK with assistance from M3C, BMK, SK, and ARA. Sample processing in Guinea was led by RT, TT, YH, JAB, and PM with help from FRK, KiS, AV, VV, BA, LO, AB, SD, and EN. Analysis of samples in the UK was done by RT, TT, YH, and KSR. Processing of whole virus ELISA and live virus neutralisation was led by TS with help from SKF and VK. The manuscript was primarily written by RT, TT, YH, VK, M3C, SL, JM, CM-F, SB, and SG with input from all authors.
Declaration of interests
SG received grants from European Commission and German Research Foundation for the conduct of this study. All other authors declare no competing interests.
Data sharing
Acknowledgments
This work was funded by the US Food & Drug Administration (HHSF223201510104C) and Horizon 2020 EU’s EVIDENT research initiative (666100). TS received funds from the German Research Foundation (197785619/SFB 1021). CM-Z and SG were supported by the German Research Foundation (GU 883/5–1 and MU 3565/3–1). MWC is joint funded by Wellcome and the UK Department for International Development (214626/Z/18/Z). We would like to acknowledge the long-term support and commitment of Coyah and Guéckédou Ebola virus disease survivors associations and all the participants in this study. We are also extremely grateful to the Guinean authorities and members of Center for Training and Research on Priority Diseases including Malaria in Guinea, including Saidou Kouyate, Abdoulaye Barry, and Ousmane Soumah for their support of this study. We would also like to acknowledge the tremendous cooperation of the personnel of the Coyah and Guéckédou Prefectural Health Directorates and Offices of the regional Prefects.
Supplementary Materials
References
- 1.
How Ebola and Marburg viruses battle the immune system.
Nat Rev Immunol. 2007; 7: 556-567
- 2.
Protective cytotoxic T-cell responses induced by Venezuelan equine encephalitis virus replicons expressing Ebola virus proteins.
J Virol. 2005; 79: 14189-14196
- 3.
Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection.
J Immunol. 2005; 175: 1184-1191
- 4.
Correlates of immunity to filovirus infection.
Viruses. 2011; 3: 982-1000
- 5.
A monovalent chimpanzee adenovirus Ebola vaccine boosted with MVA.
N Engl J Med. 2016; 374: 1635-1646
- 6.
Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge.
Nat Med. 2014; 20: 1126-1129
- 7.
Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial.
Lancet. 2015; 386: 857-866
- 8.
Ebola and hantaviruses.
FEMS Immunol Med Microbiol. 1997; 18: 281-289
- 9.
Evaluation of convalescent plasma for Ebola virus disease in Guinea.
N Engl J Med. 2016; 374: 33-42
- 10.
Human asymptomatic Ebola infection and strong inflammatory response.
Lancet. 2000; 355: 2210-2215
- 11.
Ebola virus persistence in breast milk after no reported illness: a likely source of virus transmission from mother to child.
Clin Infect Dis. 2017; 64: 513-516
- 12.
How severe and prevalent are Ebola and Marburg viruses? A systematic review and meta-analysis of the case fatality rates and seroprevalence.
BMC Infect Dis. 2016; 16: 708
- 13.
Serologic evidence of Ebola virus infection in a population with no history of outbreaks in the Democratic Republic of the Congo.
J Infect Dis. 2018; 217: 529-537
- 14.
A replication defective recombinant Ad5 vaccine expressing Ebola virus GP is safe and immunogenic in healthy adults.
Vaccine. 2010; 29: 304-313
- 15.
Development of an antibody capture ELISA using inactivated Ebola Zaire Makona virus.
Med Microbiol Immunol (Berl). 2016; 205: 173-183
- 17.
Origin and differentiation of human memory CD8 T cells after vaccination.
Nature. 2017; 552: 362-367
- 18.
Immunology and evolvement of the adenovirus prime, MVA boost Ebola virus vaccine.
Curr Opin Immunol. 2015; 35: 131-136
- 19.
Long-term coexistence of SARS-CoV-2 with antibody response in COVID-19 patients.
J Med Virol. 2020; 92: 1684-1689
- 20.
Persistence of Ebola virus in ocular fluid during convalescence.
N Engl J Med. 2015; 372: 2423-2427
- 21.
Persistence and clearance of Ebola virus RNA from seminal fluid of Ebola virus disease survivors: a longitudinal analysis and modelling study.
Lancet Glob Health. 2017; 5: e80-e88
- 22.
Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995.
J Infect Dis. 1999; 179: S177-S187
- 23.
Ebola virus neutralizing antibodies detectable in survivors of the Yambuku, Zaire outbreak 40 years after infection.
J Infect Dis. 2018; 217: 223-231
- 24.
Longitudinal analysis of the human B cell response to Ebola virus infection.
Cell. 2019; 177: 1566-1582
- 25.
Molecular evidence of sexual transmission of Ebola virus.
N Engl J Med. 2015; 373: 2448-2454
- 26.
A human memory T cell subset with stem cell-like properties.
Nat Med. 2011; 17: 1290-1297
- 27.
Ebola-specific CD8+ and CD4+ T cell responses in Sierra Leonean Ebola virus survivors with and without post viral sequelae.
J Infect Dis. 2020; jiaa268
- 28.
The ongoing evolution of antibody-based treatments for Ebola virus infection.
Immunotherapy. 2017; 9: 435-450
- 29.
Evaluation of convalescent whole blood for treating Ebola virus disease in Freetown, Sierra Leone.
J Infect. 2017; 74: 302-309
- 30.
Evidence for occurrence of filovirus antibodies in humans and imported monkeys: do subclinical filovirus infections occur worldwide?.
Med Microbiol Immunol (Berl). 1992; 181: 43-55
- 31.
Asymptomatic infection and unrecognised Ebola virus disease in Ebola-affected households in Sierra Leone: a cross-sectional study using a new non-invasive assay for antibodies to Ebola virus.
Lancet Infect Dis. 2017; 17: 645-653
- 32.
Prevalence of infection among asymptomatic and paucisymptomatic contact persons exposed to Ebola virus in Guinea: a retrospective, cross-sectional observational study.
Lancet Infect Dis. 2019; 19: 308-316
Uncited References
- 16.
Phase 1 trials of rVSV Ebola vaccine in Africa and Europe.
N Engl J Med. 2016; 374: 1647-1660
Article Info
Publication History
Published: October 13, 2020
Identification
Copyright
© 2020 Elsevier Ltd. All rights reserved.







{kind=link}